
ABINIT est un logiciel open-source distribué sous licence GNU-GPL qui permet de calculer les propriétés d’un système constitué de noyaux atomiques et d’électrons dans le cadre de la Théorie de la Fonctionnelle de la Densité (DFT), théorie ab initio, ne nécessitant pas de paramétrage sur des expériences. Il s’agit d’un projet collaboratif international [1] impliquant de nombreux développeurs en France (1), Belgique (2,3), Canada (4,5), Espagne (6), USA (7), etc. Le Laboratoire Matière en Conditions Extrêmes en est l’un des principaux contributeurs. Il est engagé, depuis maintenant plus de 20 ans, dans le développement du code.
ABINIT utilise une base d’ondes planes pour représenter les fonctions d’ondes électroniques, ne faisant aucune approximation a priori sur la nature des matériaux à traiter. Il est constitué de plusieurs modules, interagissant les uns avec les autres, implémentant chacun un formalisme dérivé de la DFT. Le code permet, avant tout, de réaliser des calculs d’état fondamental des électrons, donnant accès à l’énergie totale, les forces et les contraintes du système étudié. Ces grandeurs sont alors exploitées dans des études statiques (positions des noyaux atomiques fixées), mais aussi dynamiques, pour déduire diverses propriétés thermodynamiques de la matière.
ABINIT peut aussi calculer les réponses d’un système à différentes perturbations, donnant ainsi accès à diverses propriétés comme les spectres de vibrations, les constantes élastiques ou le tenseur diélectrique. Un module intègre aussi la théorie de perturbation des systèmes à plusieurs corps (MBPT) qui est utilisée pour obtenir les états excités de la matière. La Théorie du Champ Moyen Dynamique » (DMFT) a également été implémentée au LMCE ; il s’agit d’une extension de la DFT utilisée pour prendre en compte des effets de corrélation entre électrons.
Des membres du LMCE sont investis dans de nombreux développements de ABINIT et plus particulièrement : le comportement du code sur calculateur hautes performances [2], le formalisme de la méthode “Projector Augmented-Wave” (PAW) [3], la prise en compte des corrélations électroniques (méthodes DFT+U et DMFT) [4], les calculs de fonction de réponse par perturbation de la DFT (DFPT) [5], les calculs d’états excités en DFT dépendant du temps (TDDFT), les calculs de spectroscopie ou encore la réponse en température des matériaux.
Le champ d’application du code ABINIT au LMCE est multiple. Il s’agit, d’une part, de prévoir le comportement microscopique de la matière dans les domaines inaccessibles à l’expérience (et particulièrement la matière sous conditions extrêmes de température et de pression) et, d’autre part, de confronter les calculs à des expériences dites « élémentaires », afin d’obtenir une validation mutuelle des modèles théoriques et des dispositifs expérimentaux et ainsi d’avoir une compréhension des phénomènes microscopiques. En particulier, citons : le calcul d’équations d’états de la matière du solide à l’état plasma, la détermination de la structure atomique de matériaux soumis à de fortes sollicitations, l’étude de la diffusion d’une espèce chimique dans un solide, la détermination de diagrammes de phases (dont les transitions solide-liquide), …
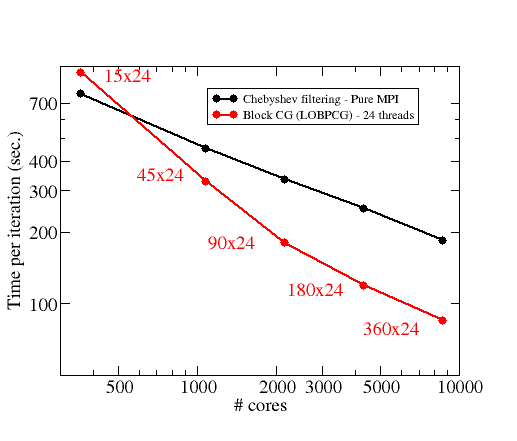
Figure 1:
Courbe de scalabilité du code ABINIT en fonction du nombre de coeurs CPU utilisés. Courbe noire: algorithme de filtrage de Chebyshev en parallélisation MPI. Courbe rouge : algorithme LOBPCG (gradient conjugé par block) en parallélisation hybride MPI+openMP. Cas test : 1960 atomes d’oxyde de Gallium (8700 états électroniques).
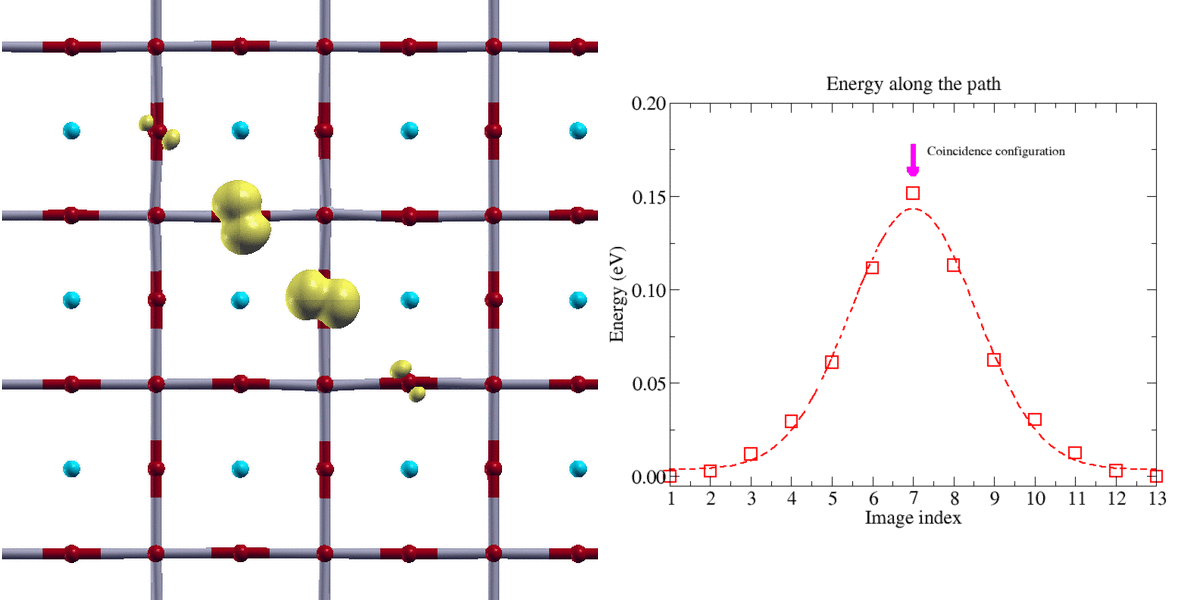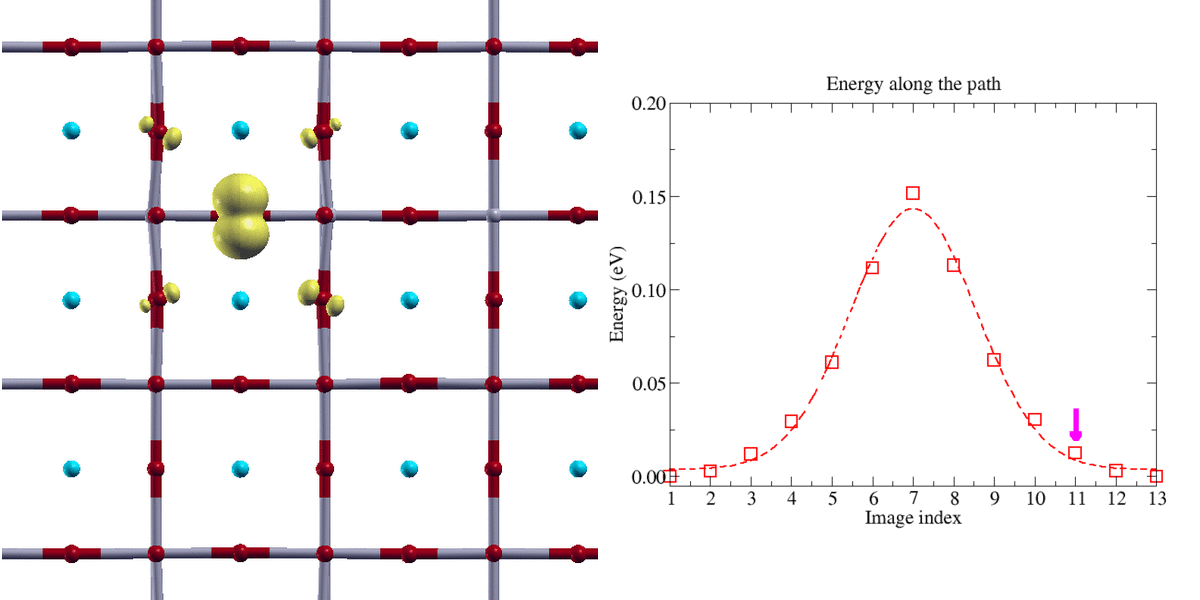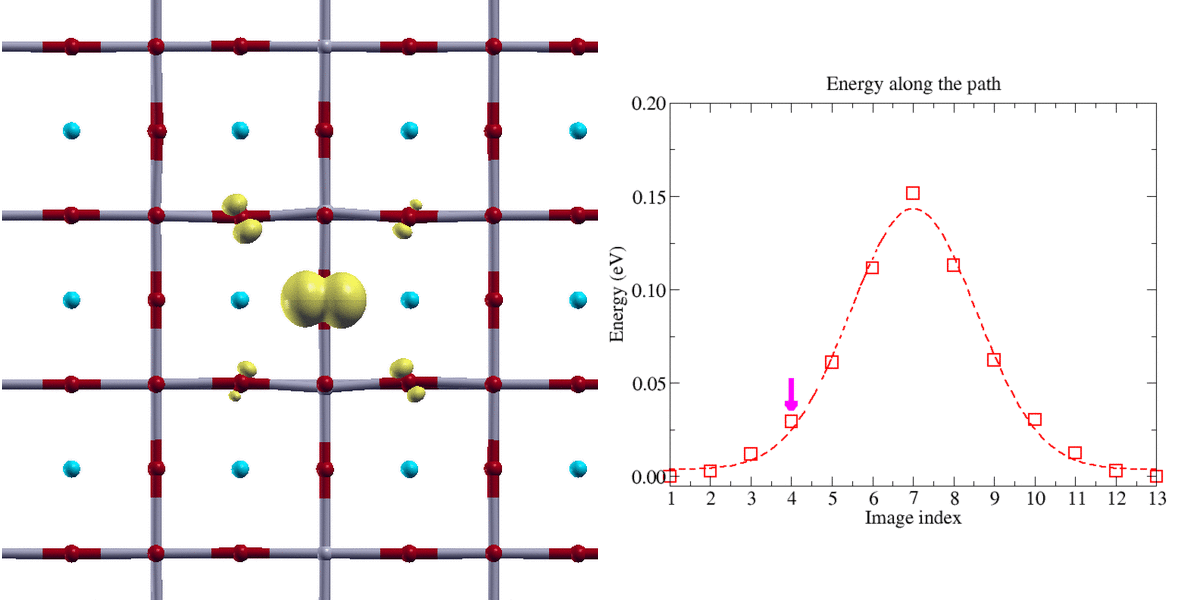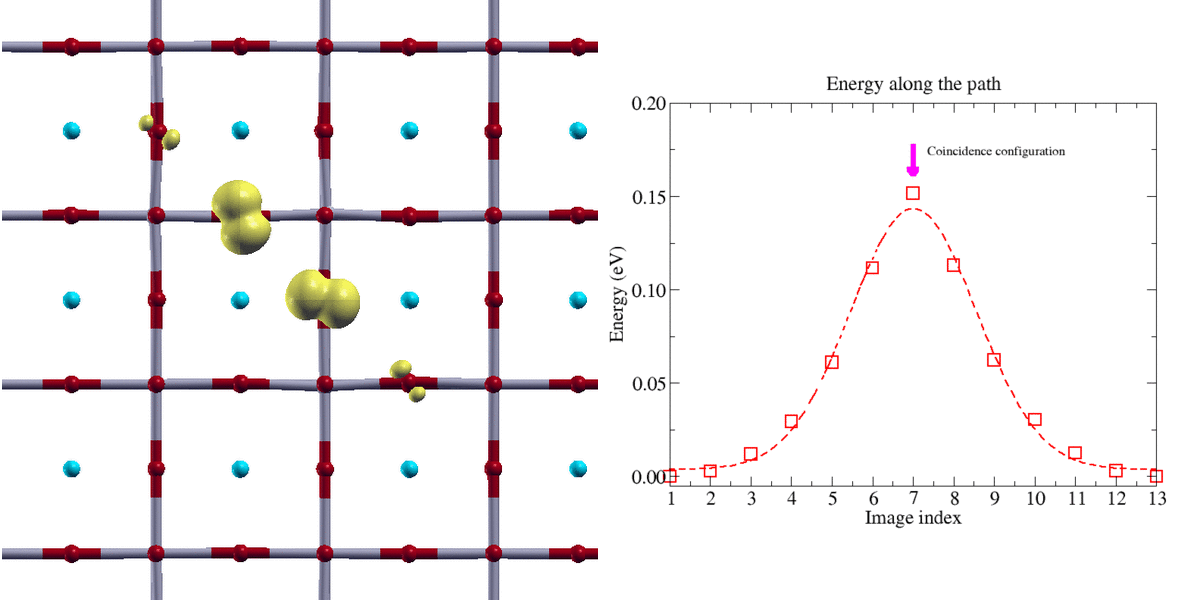
Figure 2:
Transfert adiabatique d’un polaron trou de type oxygène dans une perovskyte BaSnO3 (isolant de transfert de charge). La localisation de la charge du polaron nécessite l’emploi de la technique DFT+U. Le chemin d’énergie minimale a été obtenu avec la méthode Nudge Elastic Band (NEB)
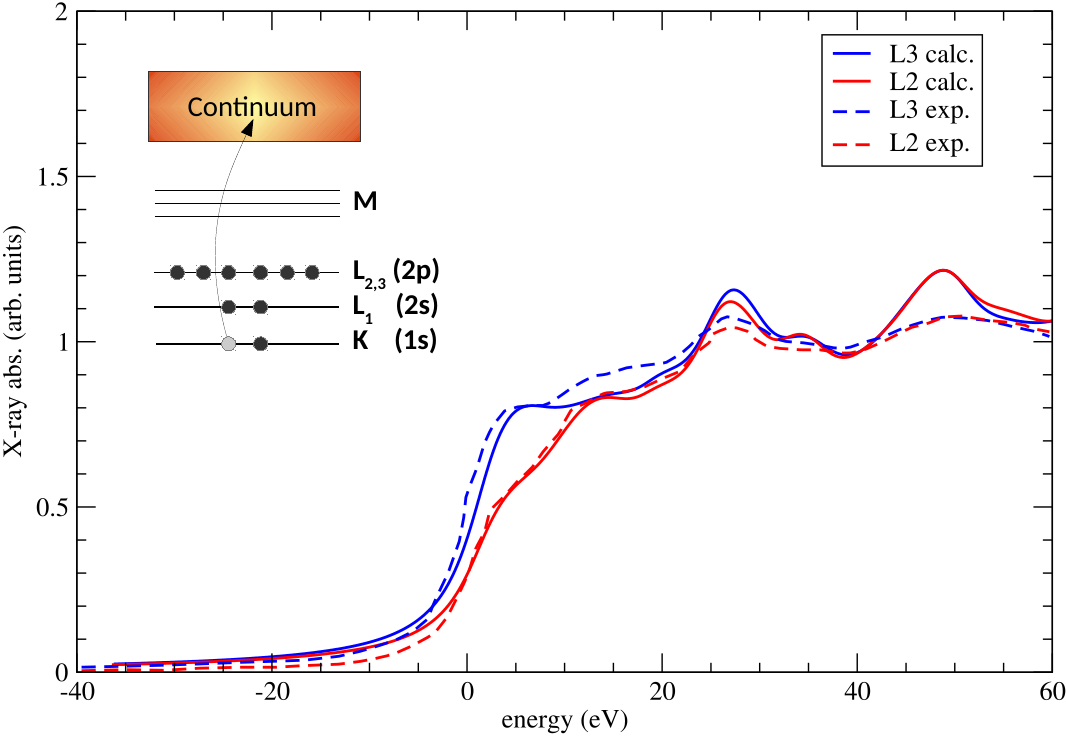
Figure 3:
Spectres de structure près du seuil d’absorption de rayons X (XANES) de l’or. La prise en compte du couplage spin-orbite pour les calculs faits avec ABINIT (traits pleins) est nécessaire pour obtenir un bon accord avec l’expérience (pointillés). Les couleurs indiquent deux types de transitions électroniques.
Publications
- A. H. Romero, D. C. Allan, B. Amadon, G. Antonius, T. Applencourt, L. Baguet, J. Bieder, F. Bottin, J. Bouchet, E. Bousquet, F. Bruneval, G. Brunin, D. Caliste, M. Côté, J. Denier, C. Dreyer, P. Ghosez, M. Giantomassi, Y. Gillet, O. Gingras, D. R. Hamann, G. Hautier, F. Jollet, G. Jomard, A. Martin, H. P. C. Miranda, F. Naccarato, G. Petretto, N. A. Pike, V. Planes, S. Prokhorenko, T. Rangel, F. Ricci, G.-M. Rignanese, M. Royo, M. Stengel, M. Torrent, M. J. van Setten, B. Van Troeye, M. J. Verstraete, J. Wiktor, J. W. Zwanziger, X. Gonze, “ABINIT: Overview and focus on selected capabilities”, J. Chem. Phys., 152, 124102 (2020) DOI
- A. Levitt, M.Torrent, “Parallel eigensolvers in plane-wave Density Functional Theory”, Comp. Phys. Comm., 187, 98 (2015) DOI
- M. Torrent, F. Jollet, F. Bottin, G. Zérah, X. Gonze, “Implementation of the projector augmented-wave method in the ABINIT code: Application to the study of iron under pressure”, Comp. Mat. Sci., 42, 337 (2008) DOI
- B. Amadon, “First-principles DFT+DMFT calculations of structural properties of actinides: Role of Hund’s exchange, spin-orbit coupling, and crystal stucture”, Phys. Rev. B, 94, 115148 (2016) DOI
- A. Martin, M. Torrent, R. Caracas, “Projector Augmented-Wave Formulation of Response to Strain and Electric-Field Perturbation within Density Functional Perturbation Theory”, Phys. Rev. B, 99, 094112 (2019) DOI
Chercheurs impliqués
B. Amadon, L. Baguet, R. Béjaud, A. Blanchet, F. Bottin, F. Brieuc, F. Gendron, G. Geneste, F. Jollet, V. Recoules, M. Torrent