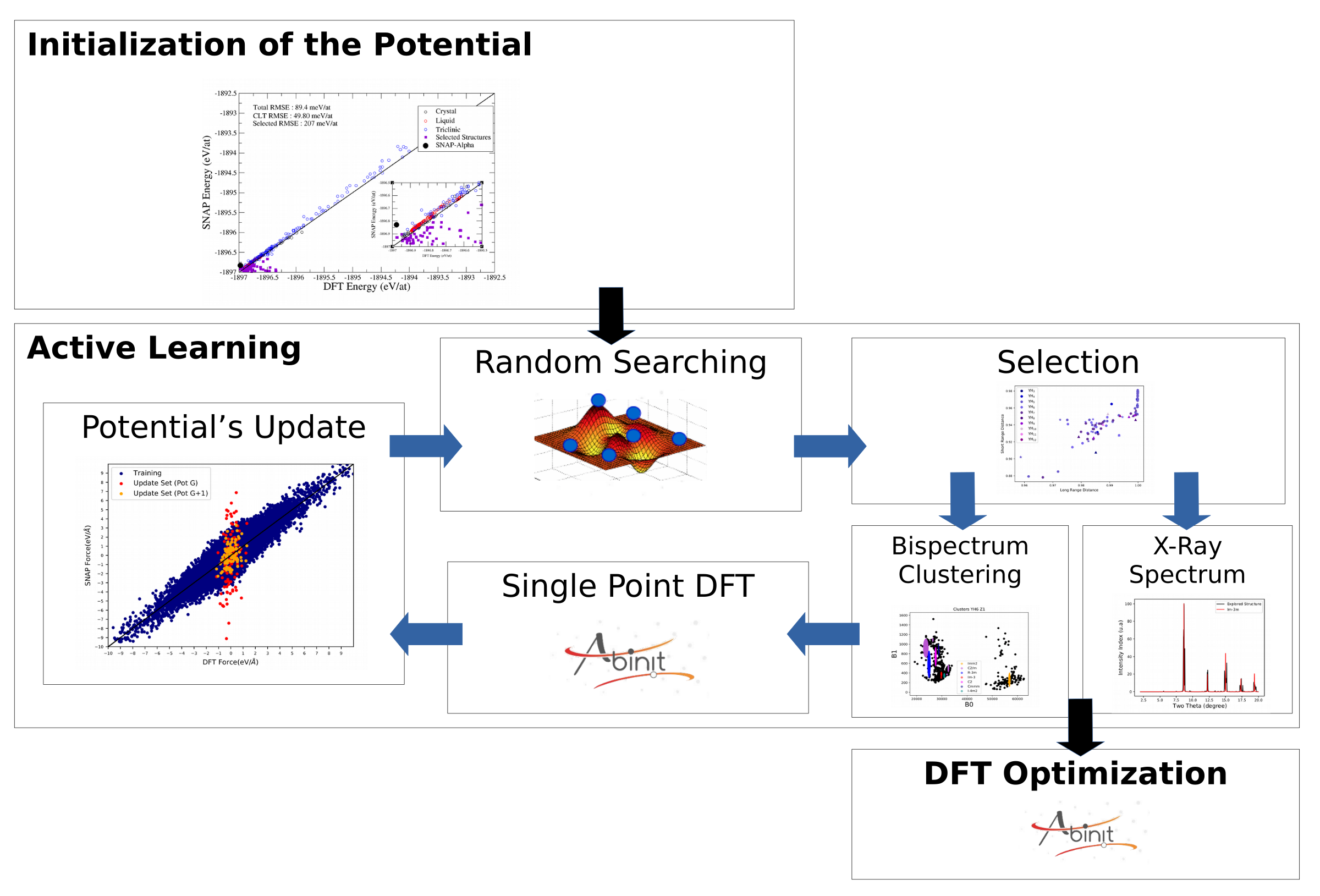
Notre recherche se concentre sur le développement de méthodologies de simulation atomistique et moléculaire prédictives pour décrire les matériaux à l’aide d’outils d’apprentissage automatique. Afin d’améliorer la précision et la transférabilité des potentiels interatomiques, nous développons des potentiels numériques basés sur des méthodes d’apprentissage automatique (SNAP, GAP, NNP) pour la description de différents phénomènes et matériaux comme, par exemple, pour étudier la structure des superhydrures [1] ou bien décrire la plasticité et les transitions de phases [2] ainsi que les contributions magnétiques [3] dans le fer. Les outils d’apprentissage automatique sont également utilisés pour caractériser les défauts dans les solides cristallins [4] et classer les structures et les dislocations à la volée. D’un point de vue plus fondamental, nous avons appliqué des réseaux neuronaux inversibles pour résoudre des problèmes inverses et déterminer le chemin d’énergie minimum associé à une transition de phase [5]. Le lien entre les simulations atomistiques et la modélisation mésoscopique des systèmes chimiquement réactifs est aussi réalisé à l’aide de techniques de ML non supervisées. Enfin, dans le but d’accélérer les simulations de type dynamique moléculaire ab initio dont le coût en temps de calcul est particulièrement élevé, nous développons l’approche Machine Learning Assisted Canonical Sampling (MLACS) [6]. Cette méthode est basée sur l’ajustement à la volée de potentiels interatomiques machine learning et permet d’accélérer considérablement l’échantillonnage de l’espace des configurations tout en gardant la précision ab initio.
Publications
- J.-B. Charraud, G. Geneste, M. Torrrent, J.-B. Maillet. “Machine learning accelerated random structure searching: Application to yttrium superhydrides” J. Chem. Phys., 156, 204102 (2022) DOI
- J.-B Maillet, C. Denoual, G. Csanyi. “Machine-learning based potential for iron: Plasticity and phase transition” AIP Conference Proceedings, 1979, 050011 (2018) DOI
- S. Nikolov, M. A. Wood, A. Cangi, J.-B. Maillet, M.-C. Marinica, A.P. Thompson, M.P. Desjarlais, J. Tranchida. “Data-driven magneto-elastic predictions with scalable classical spin-lattice dynamics” npj Computational Materials, 7, 153, (2021) DOI
- A.M. Goryaeva, C. Lapointe, A. Cangi, C. Dai, J. Dérès, J.-B. Maillet, and M.-C. Marinica “Reinforcing materials modelling by encoding the structures of defects in crystalline solids into distortion scores” Nat. Comm., 11, 4691, (2020) DOI
- M. Ramil, C. Boudier, A.M. Goryaeva, M.-C. Marinica, and J.-B. Maillet. “On Sampling Minimum Energy Path” J. Chem. Theory Comput., 18, 5864 (2022) DOI
- A. Castellano, F. Bottin, J. Bouchet, A. Levitt, G. Stoltz, “Ab initio canonical sampling based on variational inference”, Phys. Rev. B, 106, L161110 (2022) DOI
- A. Castellano, R. Béjaud, P. Richard, O. Nadeau, C. Duval, G. Geneste, G. Antonius, J. Bouchet, A. Levitt, G. Stoltz, F. Bottin “Machine Learning Assisted Canonical Sampling (MLACS)”, arXiv:2412.15370 DOI (2024)
Chercheurs impliqués
R. Béjaud, F. Bottin, F. Brieuc, G. Geneste, G. Kluth, P. Lafourcade, J.-B. Maillet, M. Torrent